Annexes au rapport de stage de Alexandre Alapetite à l'INRA.CBGP, janvier 2002
Annexes rangées par ordre d'intérêt
Notions de biologie complémentaires
Je fais ici une brève description de quelques notions et techniques très utilisées en
génétique des populations. Elles concernent mon stage car les données traitées par GeneClass 2
proviennent directement de ses manipulations, et ne peuvent être vraiment comprises qu'avec
un minimum de connaissances sur leur origine et leur signification.
Marqueurs : exemple, les microsatellites
Un marqueur génétique est un caractère mesurable à hérédité Mendélienne.
On oppose traditionnellement les marqueurs morphologiques (couleur, forme,...)
aux marqueurs moléculaires (au niveau de l'ADN) et aux marqueurs biochimiques (protéines,...).
Swynghedauw, 2000.
Les marqueurs microsatellites sont des marqueurs moléculaires souvent utilisés pour créer
les données traitées par GeneClass 2. Ils sont transmis de génération en génération,
présentent un polymorphisme élevé (taux de mutation assez fort), ne sont pas source de sélection naturelle,
et ont d'autres avantages souhaitables pour être retenus comme marqueurs.
Ce sont de courtes séquences de motifs d'ADN répétées en tandem, constituées d'unités de
répétition de longueur variant entre 1 et 6 paires de bases (pb).
Les régions flanquantes, situées de part et d'autre d'un microsatellite, permettent de le repérer.
La longueur totale d'une séquence (microsatellite et régions flanquantes) est généralement
comprise entre 10 et 200pb.
Les microsatellites les plus courants (ex: ATATATAT) sont constitués de mono [ex: (A)n]
et de dinucléotides [ex:(AT)n].
Parmi les autres types marqueurs de marqueurs utilisés, on trouvera classiquement les
isoenzymes, qui sont exprimées par des différences de charges des protéines codées
à partir de l'ADN.
Amplification : exemple, la PCR
La PCR ou Polymerase Chain Reaction permet de cibler un segment d'ADN particulier dans le génome,
puis de recopier ce segment des millions de fois et de le rendre analysable.
Dans le cas d'un locus microsatellite, le segment intéressant est limité par ses régions
flanquantes, qui le caractérisent. Ces régions flanquantes sont propres à une population ;
mais si elles sont inconnues, elles doivent être préalablement cherchées par des techniques
de "criblage", longues et coûteuses, qui sortent du cadre de ce rapport.
Nous allons voir ici les principales étapes d'un cycle de PCR.
Plusieurs dizaines de cycles doivent être effectués (en général entre 25 et 35).
 |
Présentation extraite de
http://www.ens-lyon.fr/RELIE/PCR/
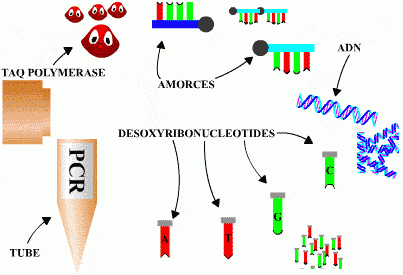
Avant la réaction, tous les acteurs de la PCR sont introduits dans le même tube.
Il s'agit de l'ADN à amplifier, des amorces pour les régions flanquantes ciblées,
de l'ADN polymérase (ici la Taq polymérase) et enfin du mélange des quatre
désoxyribonucléotides constitutifs de l'ADN (A,C,T,G).
Tous sont ajoutés en large excès par rapport à l'ADN.
|
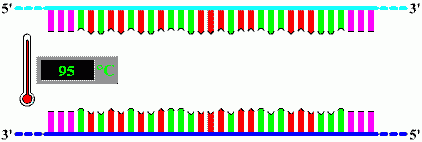 |
Dénaturation
Le thermomètre indique la température qui règne dans le thermocycleur, appareil qui permet
d'automatiser la PCR.
A cette température, les liaisons faibles qui assuraient la cohésion
de la double hélice d'ADN sont rompues pour donner deux simples brins d'ADN.
|
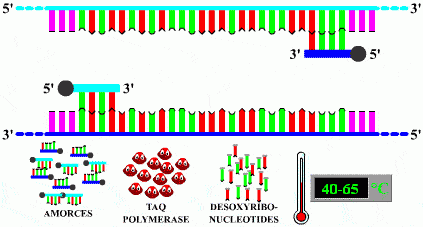 |
Hybridation
L'hybridation des amorces sur l'ADN repose sur le principe de l'appariement des bases complémentaires.
On voit les amorces en bleu foncé et bleu clair qui se sont fixées.
|
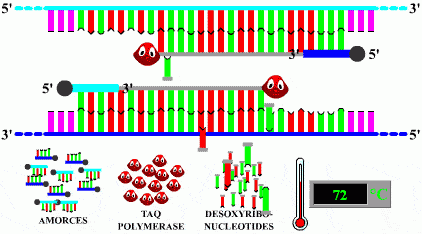 |
Élongation (extension des amorces)
Les amorces hybridées à l'ADN servent de point de départ, à la polymérisation du brin d'ADN
complémentaire de l'ADN matrice.
La polymérisation se fait par ajout successif des désoxyribonucléotides.
Chaque base ajoutée est complémentaire de la base correspondante du brin matrice.
Les ADN polymérases sont des enzymes qui synthétisent l'ADN de l'extrémité 5-prime vers l'extrémité 3-prime.
Les polymérases (ici le petit personnage rouge) utilisées en PCR sont extraites de bactéries vivant naturellement à des températures élevées.
|
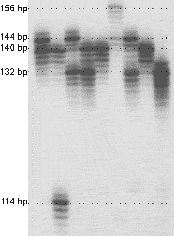
Génotypage des individus
Une fois la PCR validée sur un petit nombre d'individus, le typage des individus à l'échelle
des populations peut être réalisé, avec une PCR radioactive (isotope radioactif du nucléotide A).
- Les nombreux fragments amplifiés obtenus sont ensuite déposés sur un gel d'acrylamide, aux côtés de témoins de taille.
- La distance de migration des fragments par électrophorèse sera d'autant plus importante que les fragments sont courts.
- Le gel (pour plusieurs individus à la fois) est révélé grâce à un film radio-sensible.
- Le but est de trouver la taille du site microsatellite de chaque individu.
- La taille de ce site détermine l'allèle de l'individu pour un locus.
- La lecture du gel permet aussi de savoir si l'individu est homozygote ou hétérozygote sur le locus testé.
- Sources non-Internet :
- Anne Guichard, INRA.CBGP 1, 2001
Programmation sur la structure de données du composant TPGDSStructure
Le composant TPGDSStructure est une structure efficace, assez complète pour pouvoir traiter différents types de données de
génétique des populations, comme la ploïdie variable, fournissant assez d'informations pour pouvoir renseigner les différents algorithmes d'assignation,
comme les fréquences alléliques, la longueur des allèles.
Elle est donc un peu complexe, mais son utilisation est assez simple si l'on reste rigoureux.
Elle est basée sur l'héritage et le polymorphisme objet et nous allons voir ici rapidement comment utiliser ses fonctionnalités les plus usitées.
Voir la documentation automatique DelphiDoc.
Voici déjà l'arborescence des principaux objets (cliquez pour les méthodes et propriétés) :
TBioAbstract : Objet abstrait ayant un attribut nom.TBioCalculable : Objet abstrait sur lequel reposent tous les algorithmes généralisés : synthétise les données génétiques pour un ensemble de 1 à n individus.TLocus : Représente un locus avec ses différents allèles.TIndividual : C'est un individu avec ses caractéristiques.TGroupAbstract : Objet abstrait regroupant des individus dans un TVectorIndividuals.TGroup : Contient un ensemble TVectorGroups de groupes et/ou de populations. (noeud dans une arborescence de TGroupAbstract).TSample : C'est une population. (feuille dans une arborescence de TGroupAbstract).TVectorBioAbstract : Tableau abstrait stockant des TBioAbstract, avec toutes les méthodes nécessaires pour les gérer (ajout, suppression, recherche, ...).TVectorLoci : Spécification de TVectorBioAbstract pour stocker des TLocus.TVectorIndividuals : Spécification de TVectorBioAbstract pour stocker des TIndividual.TVectorGroups : Spécification de TVectorBioAbstract pour stocker des TGroupAbstract.
- Les individus
- Une
TPGDSStructure possède une table allIndividuals de type TVectorIndividuals contenant tous les individus.
- Pour accéder à cette table des individus:
aPGDSStructure.allIndividuals
- Pour connaître le nombre d'individus :
aPGDSStructure.allIndividuals.size
- Les locus et les allèles
- Elle dispose d'une table
allLoci de type TVectorLoci des locus contenant eux-mêmes tous les allèles trouvés pour l'ensemble des individus TIndividual.
- Pour accéder à cette table des locus et des allèles :
aPGDSStructure.allLoci
- Pour connaître le nombre de locus :
aPGDSStructure.allLoci.size
- Les populations
- La structure possède un super-groupe
allSamples de type TGroup contenant toutes les populations TSample.
- Pour accéder à une population :
aPGDSStructure.samples[i]
- Pour connaître le nombre de populations :
aPGDSStructure.nbSamples
- Pour accéder à toutes les fonctionnalités du super-groupe :
aPGDSStructure.allSamples
Description des sous-composants :
- Les populations ou les groupes sont gérés en partie par
TGroupAbstract
- Il contient un ensemble
TVectorIndividuals d'individus appartenant à cette population ou groupe.
- Pour accéder à ce tableau d'individus :
aGroupAbstract.allIndividuals
TBioCalculable gère tout ce sur quoi on peut faire des calculs
- C'est un des objets les plus importants. Il permet de généraliser les algorithmes travaillant à l'assignation d'individus, de populations et même de groupes de populations.
- Plusieurs de ses méthodes sont abstraites pour profiter du polymorphisme et ne sont réellement implémentées que dans ses descendants.
- Il précalcule et stocke des informations génétiques comme la proportion d'hétérozygotes, la diversité génétique (Nei's Gene Diversity), les fréquences alléliques, le nombre de gènes et d'allèles différents sur un locus, ...
- Pour accéder à la fréquence d'un allèle dont on connaît la référence sur un locus donné :
aBioCalculable.freqAlleleRef[iLocus,refAllele]
Logiques de programmation :
Deux points sont essentiels :
- Les locus sont toujours tous présents et dans le même ordre pour tous les individus, populations et groupes d'une même
TPGDSStructure. La gestion des données manquantes est assurée.
- Il n'en est pas de même pour les allèles, qui ne sont pas tous présents, et pas forcément dans le même ordre pour les différentes entités d'une
TPGDSStructure.
Aussi, pour comparer les fréquences d'un alléle sur un locus entre deux individus, il faudra connaître la référence de l'allèle (identifiant unique pour toute une TPGDSStructure).
Pour cela, il faut demander la référence de l'allèle dont on connaît le nom au niveau de la table des locus de la TPGDSStructure :
refAllele:=aPGDSStructure.allLoci.locus[numLocus].indexOfAllele('138');
Comme on le voit, la référence de l'allèle est en fait sa position au niveau du tableau des locus général de la TPGDSStructure.
A présent on peut accéder à la fréquence de cet allèle précis (si l'allèle n'est pas présent, sa fréquence est de 0), pour un individu, une population ou un groupe.
frequence138:=aBioCalculable.freqAlleleRef[numLocus,refAllele];
De même pour le nombre d'occurrences d'un allèle (0 si l'allèle n'est pas présent), surtout pour une population ou un groupe :
occurences138:=aBioCalculable.nbGenesRef[numLocus,refAllele];
Pour connaître le nombre d'allèles recensés dans toute la TPGDSStructure sur un locus :
aPGDSStructure.allLoci.locus[numLocus].nbAlleles;
Pour faire un parcours exhaustif des allèles d'un individu, d'une population ou d'un groupe, on peut se passer de leurs références.
Comme on le voit, le parcours des locus ne nécessite pas l'accès à la table générale des locus, vu que tous les locus sont systématiquement
présents pour toutes les entités.
for numLocus:=0 to aBioCalculable.nbLocus-1 do
begin
nbGenes:=aBioCalculable.nbGenestotal[numLocus];
for numAllele:=0 to aBioCalculable.nbAlleles[numLocus]-1 do
begin
freq:=aBioCalculable.freqAllele[numLocus,numAllele];
nbAlleles:=aBioCalculable.nbGenes[numLocus,numAllele];
...
end;
end;
L'accès aux différentes populations est aussi très utilisé. Un raccourci pour accéder aux populations a été mis en place.
for numPop:=0 to aPGDSStructureRef.nbSamples-1 do
begin
aSample:=aPGDSStructure.samples[numPop];
...
end;
On a parlé de raccourci car l'appel complet pour accéder à la structure qui gère toutes les populations est plus long mais offre plus de fonctionnalités.
Ce type d'accès est surtout utilisé par les parseurs de fichiers en interne et ne devrait pas être souvent utilisés par les utilisateurs du composant TPGDSStructure.
En fait, une TPGDSStructure contient un TGroup nommé allSamples qui contient des populations TSample.
aGroupAbstract:=aPGDSStructure.allGroups.groups.groupNamed['population1'];
if Assigned(aGroupAbstract) then
aSample:=TSample(aGroupAbstract);
...
aPGDSStructure.allGroups.groups.addGroup(aSample);
TMatchPGDS, une structure de croisement
La particularité des algorithmes d'assignation est qu'ils utilisent deux structures TPGDSStructure, une servant de référence,
l'autre contenant les échantillons à assigner. Pour faciliter et optimiser le croisement de deux TPGDSStructure, une
structure intermédiaire permettant une interaction simple et efficace a été créée. Voici le modèle d'utilisation le plus courant :
aMatchPGDS:=TMatchPGDS.create(PGDSStructureRef,PGDSStructureSamp,listeLocusUtiles);
for iLocusCurrent:=0 to matchPGDS.nbLoci-1 do
with aMatchPGDS.matchLoci[iLocusCurrent]^ do
begin
for hAllele:=0 to Pred(nbAllelesSamp+nbAllelesRef) do
with matchAlleles[hAllele] do
begin
end;
end;
aMatchPGDS.Free();
Assignation Baudouin, exemple d'algorithme d'assignation
Voici un exemple d'algorithme généralisé, qui permet de faire l'assignation Baudouin ou Rannala pour des individus ou des populations.
On remarquera surtout l'utilisation de deux structures de données dataStructRef,dataStructSamp ainsi que de la
structure de croisement matchPGDS.
procedure _assignationBaudouinRannala(const dataStructRef,dataStructSamp:TPGDSStructure;
const matchPGDS:TMatchPGDS; const sampleSamp:TBioCalculable;
const filtre:Single; const assignMethod:TAssignMethod; aResultatsPopulation:TResultats);
var
iPopRef : Integer;
iLocusCurrent : Integer;
sampleRef : TSample;
hAllele : Integer;
mh,nh : Integer;
m,n : Integer;
k : Integer;
alpha : Extended;
S1, S2, S3 : Extended;
LnP : Extended;
sommeScore : Extended;
produitScore : Extended;
begin
aResultatsPopulation.size:=dataStructRef.allSamples.groups.size;
aResultatsPopulation.nomEchantillon:=sampleSamp.treeName;
sommeScore:=0;
for iPopRef:=0 to aResultatsPopulation.size-1 do
begin
sampleRef:=dataStructRef.samples[iPopRef];
LnP := 0.0;
for iLocusCurrent := 0 to matchPGDS.nbLoci-1 do
with matchPGDS.matchLoci[iLocusCurrent]^ do
begin
k := nbAllelesSamp+nbAllelesRef;
case assignMethod of
asmBaudouin: alpha := k;
asmRannala: alpha := 1;
else alpha := k;
end;
n := sampleRef.nbGenestotal[refLocRef];
m := sampleSamp.nbGenestotal[refLocSamp];
S1 := 0.0; S2 := 0.0; S3 := 0.0;
for hAllele:=0 to Pred(k) do
begin
with matchAlleles[hAllele] do
begin
if refAlleleRef<0 then nh:=0
else nh := sampleRef.nbGenesRef[refLocRef,refAlleleRef];
if refAlleleSamp<0 then mh:=0
else mh := sampleSamp.nbGenesRef[refLocSamp,refAlleleSamp];
end;
S1 := S1 + LnGamma(mh + nh + alpha/k);
S2 := S2 + LnGamma(mh + 1);
S3 := S3 + LnGamma(nh + alpha/k);
end;
LnP := LnP + LnGamma(m + 1) + LnGamma(n + alpha) + S1 - S2 - S3 - LnGamma(m + n + alpha);
end;
with aResultatsPopulation.resultat[iPopRef]^ do
begin
valeur := Exp(LnP);
intitule := sampleRef.name;
if isNan(valeur) then valeurTri := MinExtended
else
begin
valeurTri := valeur;
sommeScore:=sommeScore+valeurTri;
end;
end;
end;
aResultatsPopulation.prepareTrieTronque();
if sommeScore>0 then produitScore:=100.0 / sommeScore
else produitScore:=100.0;
for iPopRef:=0 to aResultatsPopulation.size-1 do
with aResultatsPopulation.resultatTrieTronque[iPopRef]^ do
valeurTri:=valeurTri*produitScore;
aResultatsPopulation.TrierTronquer(filtre);
end;
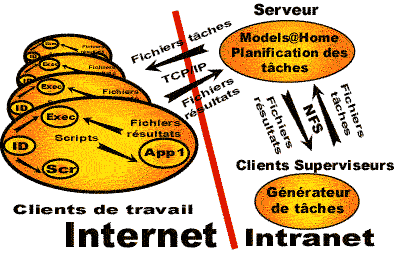
Utilisation du clustering avec Models@Home |
|
 |
 |
|
Les transferts de données et d'ordres dans un cluster Models@Home se font essentiellement par fichiers.
- ID : Détecteur d'inactivité pour démarrer les calculs gérés par un écran veille (Scr).
- Exec : Module d'exécution client. Comme l'écran de veille (Scr) mais en permanence.
- App1 : L'application appelée pour faire le calcul. Cette application peut être transmise par le serveur.
Il y a donc un serveur, plusieurs clients (ex:10), et en général un seul superviseur.
|
Le superviseur écrit (par NFS) dans un fichier que le serveur lit en permanence.
Il peut aussi transmettre les applications qui feront les calculs chez les clients.
Le superviseur doit détecter ensuite les résultats qu'il a commandés, qui arrivent sous forment de fichiers textes, et en faire la synthèse.
Nous avons écrit un superviseur graphique, en Delphi, qui fonctionne aussi bien sous Windows que sous Linux.
Chez les clients, les versions consoles de GeneClass 2 sont utilisées.
Ce cluster permettra de faire de tests de puissances des algorithmes d'assignation de GeneClass 2.
Exemple de parseur : format Cirad Baudouin PRN
Voici un exemple de parseur en Lex & Yacc pour Delphi. Les sources Lex & Yacc sont présentées ici à titre indicatif,
et ne peuvent pas être comprises telles quelles. cf choix de Lex & Yacc.
Le résultat du traitement par Lex & Yacc n'est pas montré, car il est long, peu explicite,
et sans grand intérêt (c'est un long automate à états).
Source Lex pour format Baudouin
Le fichier a été modifié pour des raisons de mise en page. Des fins d'expressions ont été passées à la ligne,
ce qui n'est pas forcément valide pour Lex.
Une fois traité par Lex, on obtient ciradprnlex.inc, un morceau de fichier source Delphi.
%{
%}
%START INITIAL xPOPULATIONS xPOPULATION xINDIVIDUAL
xINITIALCOMMENT xPOPULATIONSCOMMENT xPOPULATIONCOMMENT xINDIVIDUALCOMMENT
eol (\r\n)|(\n\r)|\r|\n
spaces [ \t]+
comment \*
pop ^\{[^\r\n\*\{\}]{1,6}
endpop ^\}
nom [^\*\r\n\t\{\} ]{1,4}
allele [0-9]{3}
allelenul [nN][iuIU][lL]
%%
<INITIAL,xPOPULATIONS,xPOPULATION,xINDIVIDUAL>{spaces} begin end;
<INITIAL,xPOPULATIONS,xPOPULATION>{eol} begin progress(); end;
<INITIAL>{comment} begin start(xINITIALCOMMENT); end;
<xINITIALCOMMENT>[^\r\n]* begin end;
<xINITIALCOMMENT>{eol} begin progress(); start(INITIAL); end;
<INITIAL>{nom} begin yylval:=yytext; if yylval='DATA' then
begin start(xPOPULATIONS); return(tokDATA); end
else return(tokLOCUS); end;
<xPOPULATIONS>{comment} begin start(xPOPULATIONSCOMMENT); end;
<xPOPULATIONSCOMMENT>[^\r\n]* begin end;
<xPOPULATIONSCOMMENT>{eol} begin progress(); start(xPOPULATIONS); end;
<xPOPULATIONS>{pop} begin yylval:=yytext; start(xPOPULATION); return(tokSTARTPOPULATION); end;
<xPOPULATION>{comment} begin start(xPOPULATIONCOMMENT); end;
<xPOPULATIONCOMMENT>[^\r\n]* begin end;
<xPOPULATIONCOMMENT>{eol} begin progress(); start(xPOPULATION); end;
<xPOPULATION>{nom} begin yylval:=yytext; start(xINDIVIDUAL); return(tokSTARTINDIVIDUAL); end;
<xPOPULATION>{endpop} begin start(xPOPULATIONS); return(tokENDPOPULATION); end;
<xINDIVIDUAL>{allele} begin yylval:=yytext; return(tokALLELE); end;
<xINDIVIDUAL>{allelenul} begin yylval:=yytext; return(tokALLELENUL); end;
<xINDIVIDUAL>{eol} begin progress(); start(xPOPULATION); return(tokENDINDIVIDUAL); end;
<xINDIVIDUAL>{comment} begin start(xINDIVIDUALCOMMENT); end;
<xINDIVIDUALCOMMENT>[^\r\n]* begin end;
<xINDIVIDUALCOMMENT>{eol} begin progress(); start(xINDIVIDUAL); end;
%%
Source Yacc pour format Baudouin
Le fichier a été modifié pour des raisons de mise en page, ce qui n'est pas forcément valide pour Yacc.
Notez la ligne {$INCLUDE ciradprnlex.inc} qui inclue le fichier généré par Lex dans celui généré par Yacc,
pour faire une seule unité ciradprn.pas Delphi valide, capable d'interpréter un fichier CIRAD PRN (sur lequel on a branché un flux TStream),
par l'appel de la méthode parseCiradPRN(...).
%{
unit ciradprn;
interface
uses U_TPGDSStructure, Classes;
type TYaccEvent = procedure(const progression:Integer; out stop:Boolean) of Object;
function parseCiradPRN(var dataStructure:TPGDSStructure; aStream:TStream; yaccEvent:TYaccEvent):Boolean;
function yystart():Integer;
procedure yyerror(const msg:String; state:Integer; lineno, colno:Integer);
resourcestring
GENOTYPE_MANQUANT = 'Génotype manquant';
TROP_DE_GENOTYPES = 'Trop de génotypes';
ALLELE_MANQUE = 'Génotype manquant';
ERREUR_LECTURE = 'Erreur "%s" à la ligne %d colonne %d en lecture de fichier CIRAD PRN';
implementation
uses lexlib, yacclib, U_TLocus, U_TSample, U_TIndividual, U_lib, SysUtils;
type YYSType = String;
var
DS : TPGDSStructure;
onYaccEvent : TYaccEvent;
iLoc : Integer;
iAllele : Integer;
popName : String;
a1 , a2: String;
iA : Integer;
Samp : TSample;
Ind : TIndividual;
procedure yydebugoutput(msg : String);
begin
msg:=msg+EOL;
yyoutput.Write(msg[1],Length(msg));
end;
function yywrap():Boolean;
begin
Result := True;
end;
procedure progress();
var stop:Boolean;
begin
if Assigned(onYaccEvent) then
begin
onYaccEvent(100 * yyinput.Position div yyinput.Size,stop);
if stop then
begin
yyabort();
yyerror(FILE_STOP,yystate,yylineno,yycolno);
end;
end;
end;
%}
%token
tokLOCUS
tokDATA
tokSTARTPOPULATION
tokENDPOPULATION
tokSTARTINDIVIDUAL
tokENDINDIVIDUAL
tokALLELE
tokALLELENUL
%%
Input:
Header tokDATA Populations {
DS.fileType := fbtPRN;
yyaccept();
};
Header:
tokLOCUS {
DS.allLoci.addLocus(TLocus.create($1));
} | Header tokLOCUS {
DS.allLoci.addLocus(TLocus.create($2));
};
Populations: Population | Populations Population;
Population: StartPopulation Individus tokENDPOPULATION;
StartPopulation:
tokSTARTPOPULATION {
PopName := Trim( Copy($1, 2, Length($1)-1) );
Samp := TSample.create(PopName, DS.allLoci.size);
DS.allSamples.groups.addGroup(Samp);
};
Individus: LigneIndividu | Individus LigneIndividu;
LigneIndividu:
NomIndividu Typage | LigneIndividu Typage
| LigneIndividu tokENDINDIVIDUAL {
if iAllele=1 then yyerror(ALLELE_MANQUE, yystate, yylineno, yycolno);
if iLoc < DS.allLoci.size then
yyerror(GENOTYPE_MANQUANT, yystate, yylineno, yycolno)
else if iLoc > DS.allLoci.size then
yyerror(TROP_DE_GENOTYPES, yystate, yylineno, yycolno);
};
NomIndividu:
tokSTARTINDIVIDUAL {
Ind := TIndividual.create($1, DS.allLoci.size, 2);
DS.allIndividuals.addIndividual(Ind);
Samp.allIndividuals.addIndividual(Ind);
iLoc := 0;
iAllele :=0;
};
Typage:
tokALLELE {
if (iAllele=0) then
begin
a1 := $1;
iA:=DS.allLoci.locus[iLoc].declareAlleleGlobal(a1, strToInt(a1));
Ind.addGene(iLoc, iA);
Inc(iAllele);
end
else
begin
a2 := $1;
iA:=DS.allLoci.locus[iLoc].declareAlleleGlobal(a2, strToInt(a2));
Ind.addGene(iLoc, iA);
Inc(iLoc);
iAllele:=0;
end;
} | tokALLELENUL {
if (iAllele=0) then
begin
Inc(iAllele);
end
else
begin
Inc(iLoc);
iAllele:=0;
end;
};
%%
function yystart():Integer;
begin
yylineno:=0;
yyinput.Seek(0, soFromBeginning);
start(INITIAL);
Result := 0;
end;
procedure yyerror(const msg:String; state:Integer; lineno,colno:Integer);
var vException:EFileFormatWarning;
begin
vException := EFileFormatWarning.CreateResFmt(Integer(@ERREUR_LECTURE),[msg,lineno,colno]);
DS.parseErrors.Add(vException.Message);
raise vException;
end;
function parseCiradPRN(var dataStructure:TPGDSStructure; aStream:TStream; yaccEvent:TYaccEvent):Boolean;
var trash : String;
begin
DS := dataStructure;
onYaccEvent:=yaccEvent;
yyclear();
yyinput := aStream;
yyoutput := TFileStream.create('ciradprn.txt', fmCreate or fmShareDenyWrite);
yyoutput := TStringStream.create(trash);
yystart();
Result := (yyparse() = 0);
yyoutput.Free();
end;
end.
Journal d'activité
- 1er octobre 2001
- Début du stage.
- 1/10/2001 - 03/10/2001
- Étude de la situation de départ, outils utilisés, besoins.
- 4/10/2001 - 15/10/2001
- Développement au Cirad d'une version d'essai d'un outil d'assignation des populations
- Création d'un format de données sur fichier texte, à base de XML.
- 15 octobre 2001
- Intégration à l'INRA.
- 15/10/2001 - 01/10/2001
- Création d'une structure de données permettant de stoker et de manipuler les individus, leurs populations, ainsi que leurs caractéristiques (fréquence allélique,...).
- Apprentissage des générateurs Lex & Yacc (lexical et grammatical).
- Modification de la bibliothèque de Lex pour lire les fichiers PC, Linux, MAC.
- Lecture et écriture de différents formats de fichier utilisés au CIRAD.CP et à l'INRA.CBGP par les logiciels de statistique sur les populations.
- 02/11/2001 - 07/11/2001
- Début d'interface graphique.
- Implémentation d'un algorithme d'assignation de populations (méthode Baudouin).
- 08/11/2001 - 11/11/2001
- Correction de bugs, création de routines utilitaires.
- Portage du programme sous Linux avec Kylix.
- 12/11/2001 - 13/11/2001
- Début des rapports.
- 14/11/2001 - 15/11/2001
- Étude du support de plusieurs langues (Français, Anglais, Italien, Allemand, ...)
- 16/11/2001 - 18/11/2001
- Interface graphique.
- 19/11/2001 - 20/11/2001
- Commentage et nettoyage du code source.
- Compilation sous Linux avec Kylix 2.
- Réunion à l'INRA.CBGP mardi 20 novembre de 10h00 à 11h45.
- 21/11/2001 - 22/11/2001
- Commentaire des sources, début d'une aide pour les développeurs.
- Test de l'exécutable généré par Kylix 2 sous Linux (Mandrake 8.1) utilisant successivement les environnements graphiques KDE, Gnome, AfterStep, Enlightment, SawFish passé avec succès.
- 23/11/2001 - 27/11/2001
- Mise en forme des sources de la structure de données en package CLX pour une utilisation facile par les développeurs Borland Delphi et Kylix voire C++Builder.
- 27/11/2001
- Amélioration de la gestion des erreurs de lecture des différents formats de fichiers.
- 01/12/2001 - 02/12/2001
- Création d'un outil d'installation de GeneClass 2 pour Windows avec NullSoft Install System.
- 03/12/2001 - 05/12/2001
- Détermination des fichiers à distribuer pour la version Linux.
- Vérification de la portabilité du package sous C++Builder 5, Delphi 4, Delphi 5, Kylix 1, Kylix 2.
- Choix d'un outil de documentation automatique à la mode JavaDoc : SoftConsult DelphiDoc 2.0
- Début de commentaire des sources pour DelphiDoc.
- Aide à l'intégration d'un logiciel existant (Pop100Gen) au sein du programme actuel, en créant un composant CLX réutilisable.
- 05/12/2001 - 06/12/2001
- Transformation de la structure objet de la structure de donnée principale pour uniformiser les algorithmes travaillant au niveau des individus, des populations, ou des groupes.
- 07/12/2001
- Application de l'uniformisation des algorithmes à l'assignation Baudouin, qui permet alors d'assigner des populations ou des individus.
- 10/12/2001 - 11/12/2001
- Correction de bug dans le Lex du format Cirad PRN.
- Modification du parseur XML pour plus d'évolutivité.
- Début d'implémentation du test de hors-type (Baudouin).
- Interface graphique.
- Déboguage.
- 12/12/2001 - 20/12/2001
- Aide à l'ajout de nouvelles méthodes d'assignation.
- Déboguage.
- 22/12/2001 - 24/12/2001
- Création d'une structure de croisement permettant d'optimiser et de simplifier les calculs entre 2 structures de données principales.
- Grosse optimisation du temps de calcul (plus de 65% de gain en moyenne).
- Ajout de commentaires.
- Modification de code pour être plus strict (support de la compilation avec vérification des types @).
- Résolution de problèmes en attente (TODO).
- 25/12/2001 - 17/01/2002
- Rapport de stage, aidé par Sylvain Piry.
- 03/01/2002
- Implémentation du "leave one out" pour toutes les méthodes d'assignation.
- 04/01/2002
- Déboguage.
- Améliorations.
- Commentaires.
- 07/01/2002
- Déboguage utilisation des allèles de la structure de référence.
- 08/01/2002
- Validation des calculs (avec GeneClass1 et Pop100Gene) et correction des erreurs.
- Ajout d'un menu de gestion des fenêtres.
- Variantes d'optimisation avec compilation conditionnelle (dichotomie contre séquentiel, etc...).
- Ajout d'un seuil maximum (5) pour le nombre de scores affichés avant les résultats réels.
- 09/01/2002
- Validation distance Goldstein.
- Recherche de gain et optimisation de la fonction LnGamma() qui est une fonction critique dans les calculs (jusqu'à 40% du temps de calcul).
- Mise en place d'un système multilingues, fonctionnel sous Delphi et Kylix, utilisant une partie de l'expert langue Delphi.
- Traduction anglaise presque complète.
- 10/01/2002
- Correction bug d'écriture au format Baudouin PRN
- Validation de la version anglaise sur Kylix
- Création de la version anglaise de l'installeur NSI
- Tests d'exécution sur des postes vierges (de Delphi)
- Mise en place d'un système aisé de mise à jour de l'application dans les langues prises en charge
- 11/01/2002
- Amélioration du menu fenêtres
- Mise à jour du script d'installation pour NSIS 1.94 avec compression bzip2.
- 15/01/2002
- Mise en place d'une correction des calculs dans le cas de données manquantes.
- 16/01/2002
- Développement de la version console de GeneClass 2.
- 17/01/2002
- Début de mise en place d'un système de calcul distribué.
- 18/01/2002
- Développement en Delphi d'un dispatcheur pour Model@Home (Windows/Linux).
- Correction d'un bug dans le parseur GenePop.
- 21/01/2002 - 22/01/2002
- Mise en place du clustering.
- 23/01/2002
- Préparation de la présentation de GeneClass 2 et de l'assignation au CBGP.
- 24/01/2002 - 25/01/2002
- Aide à l'intégration de
TPGDSStructure dans un autre logiciel de génétique développé par Jean-Marie Cornuet (Introgène).
- Conseils de développement et d'utilisation de Delphi à Jean-Marie Cornuet.
- Migration de Introgène terminée et réussie.
- 26/01/2002 - 27/01/2002
- Préparation de la soutenance pour la FAC.
Alexandre Alapetite