Ministère de l’Éducation Nationale
UNIVERSITÉ MONTPELLIER II
SCIENCES ET TECHNIQUES DU LANGUEDOC
IUP
GÉNIE MATHÉMATIQUE ET INFORMATIQUE
RAPPORT DE STAGE
effectué pour le
Centre de Coopération International en Recherche Agronomique pour le Développement,
département Cultures Pérennes,
au
Centre de Biologie et de Gestion des Populations,
de l’ Institut National de Recherche Agronomique
d’octobre 2001 à janvier 2002
par
Alexandre Alapetite
Directeurs de stage :
Développement d’outils informatiques permettant l’identification de
populations végétales ou animales à l’aide de marqueurs moléculaires
en utilisant l’information extraite de l’ADN
d’un petit nombre d’individus
|
Université Montpellier II
IUP Génie Mathématiques et Informatique
Case courrier 025
Place Eugène Bataillon
34095 Montpellier cedex 5
Secrétariat : +33.4.67.14.49.52
Fax : +33.4.67.14.49.53
|
|
Centre de Biologie et de Gestion des Populations
CBGP Campus International de Baillarguet
CS 30 016
34988 Montferrier-sur-lez
Tél : +33.4.99.62.33.00
Fax : +33.4.99.62.33.45
|
|
CIRAD dép Cultures Pérennes
programme Cocotier
Avenue Agropolis
34398 Montpellier cedex 5
Tél : +33.4.67.61.58.00
Fax : +33.4.67.61.59.86
|
Remerciements
Bien que je l’aie déjà fait oralement, je réitère ici de manière plus administrative quelques remerciements aux personnes sans qui ce stage n’aurait pas été possible,
ou tout du moins plus hasardeux et moins agréable.
Je tiens à remercier particulièrement Sylvain Piry, avec qui j’ai coopéré avec joie tout au long de ce stage, ce qui a permis beaucoup d’échanges constructifs.
Je remercie bien sûr Luc Baudouin, instigateur de cette ATP, ainsi que Chantal Hamelin qui m’ont recruté en tant que stagiaire puis orienté pendant un semestre.
Un grand merci à Jean-Marie Cornuet, toujours là quand il faut pour résoudre les problèmes théoriques…
D’une manière générale, je salue la confiance que tout le monde m’a portée, notamment dans le choix des outils.
Je n’oublie pas l’équipe du CBGP, qui m’a si chaleureusement accueilli, en particulier Sandrine, Florent, Karine et les stagiaires avec qui j’ai partagé un bureau (Valeria, Sabine, …).
Du côté de l’IUP, je remercie Michel Habib pour s’être occupé de mon stage et même déplacé malgré son emploi du temps chargé.
Résumé
Mon stage a consisté à développer un composant encapsulant une structure de données concernant la génétique des populations.
Un logiciel permettant d’effectuer des assignations de populations ou d’individus à des populations de références
a été conçu en utilisant cette structure de données.
Le développement a été fait pour Windows et Linux grâce au tandem Borland Delphi / Kylix.
Ce stage a abouti à un produit fonctionnel.
Les principales innovations par rapport aux solutions précédentes
sont qu’il permet l’assignation de populations complètes, et qu’il traite des individus n-ploïdes.
Le logiciel, et surtout le composant structure de données, servent de base à un projet de 3 ans (ATP),
au cours duquel des modifications, de nouvelles méthodes et idées seront ajoutées.
L’évolutivité et la réutilisabilité ont donc été des priorités.
Cela n’a pas été au détriment de la qualité de l’interface graphique,
prévue pour que utilisateurs ni statisticiens ni informaticiens puissent l’utiliser agréablement.

Sommaire
Avant propos
Introduction
Un stage en entreprise est effectué lors du premier semestre de la 3ème année à l’IUP Génie Mathématiques et Informatique de l’université de Montpellier II.
Je l’ai effectué au Centre de Biologie et de Gestion des Populations de l’INRA de Montpellier pour le département Cultures Pérennes du CIRAD de Montpellier,
dans le cadre d’une “Action Thématique Programmée” de 3 ans détaillée ci-dessous.
Bases du problème : Assignation
Les biologistes de L’INRA.CBGP et du CIRAD.CP font des calculs statistiques sur des
informations génétiques (génotypes) issues d’échantillons, qui peuvent être des ensembles
d’individus végétaux ou animaux, avec différentes caractéristiques biologiques
fournies par des marqueurs moléculaires ADN.
Grâce aux caractéristiques génétiques des individus étudiés, on établit les
caractéristiques des échantillons.
Le but général des calculs d’assignation est de déterminer quels sont les échantillons
dont les caractéristiques sont proches d’échantillons de référence, déjà connus.
On peut faire les mêmes calculs sur des individus isolés.
Notions de biologie
Chaque individu, animal ou végétal, possède un certain nombre de répétitions de son jeu
chromosomique, c’est sa ploïdie.
Ainsi, pour les individus ayant 1, 2, 3, 4,… répétitions de leur jeu chromosomique,
on parlera respectivement d’individus haploïdes, diploïdes, triploïdes, tétraploïdes, …
Un locus est une zone précise d’un chromosome donné (un fragment d’ADN),
contenant une certaine information, codée par plusieurs bases azotées A,C,T ou G.
Un individu poly-ploïde pourra donc avoir plusieurs variantes de l’information
contenue sur un même locus ; ces variantes sont appelées allèles.
Les chromosomes contiennent une infinité de locus, mais tous ne sont pas autant
techniquement utilisables, ou peuvent ne pas être pertinents pour les calculs souhaités.
On se limite en général à l’étude de 10 ou 20 locus, choisis pour le fait qu’ils sont
polymorphes (l’information qui s’y trouve varie en fonction des
mutations génétiques qui les affectent).
On parle alors de marqueurs moléculaires.
En analysant les allèles trouvés sur les locus choisis, et ce sur plusieurs individus
d’une population, on en déduit les fréquences d’apparition des différents allèles
sur les locus choisis. Ce sont les fréquences alléliques.
Deux individus distincts, même au sein d’une même population, ont une probabilité
d’être identiques pour les locus considérés d’autant plus faible que le nombre de locus
est grand, et que la variabilité de chaque locus est grande.
Néanmoins, les fréquences alléliques permettent de caractériser une population (groupe d’individus)
de manière d’autant plus précise que l’on a étudié beaucoup d’individus et surtout que le nombre de locus est important.
En pratique, on ne dispose pas d’un grand nombre d’individus par échantillon (population),
de l’ordre de 5 à 30, les fréquences alléliques des échantillons sont donc imprécises.
Voir en annexe les notions de biologie complémentaires.
Calculs statistiques
Le problème traité durant ce stage est l’assignation d’échantillons tests à des populations de référence :
- On dispose d’une base de données contenant les fréquences alléliques des populations de référence.
- On a un ensemble d’individus constituant un échantillon test pour lequel on calcule les fréquences alléliques.
- Un calcul statistique (par exemple un calcul Bayesien) permet pour chaque échantillon de trouver les populations de références desquelles ils se rapprochent le plus.
Public d’utilisateurs visé
Les publics visés par les résultats de ce stage sont :
- Les différents biologistes et chercheurs du CIRAD.CP et de l’INRA.CBGP,
afin qu’ils puissent mieux comprendre les relations de proximité génétique entre les populations qu’ils étudient.
- Les chercheurs en génétique des populations qui pourront programmer facilement de
nouvelles formules d’assignation (ou autres), les éprouver, les comparer…
- D’autres personnes intéressées par le problème : GeneClass 2 est disponible sur le site Internet du CBGP.
L’utilisation à terme peut inclure la caractérisation d’échantillons à des fins de contrôle de
traçabilité (contrôle de variétés de plantes, de races d’élevage), ou même la médecine légale.
Intégration à un milieu professionnel
Le CIRAD (Centre de Coopération Internationale en Recherche Agronomique pour le Développement)
est un Établissement Public à Caractère Industriel et Commercial (EPIC) né en 1984 spécialisé dans
la recherche agronomique appliquée aux régions chaudes.
Sa mission est de contribuer au développement rural des pays tropicaux et subtropicaux par des
actions de recherche, des réalisations expérimentales, des actions de formation (France et étranger)
ou encore la diffusion d’information scientifique et technique.
Il travaille en coopération avec plus de 90 pays d’Afrique, d’Asie, du Pacifique, d’Amérique Latine
et d’Europe.
Les différents travaux sont réalisés dans des centres qui lui sont propres, et au sein de structures
nationales de recherche agronomique des pays partenaires.
Le CIRAD emploi 1800 personnes dont la moitié sont des cadres de recherches en métropole ou à l’étranger.
Son budget annuel de 152 millions d’euros provient pour 2/3 du Budget Civil de Recherche et de Développement technologique (BCRD)
et pour 1/3 de ressources contractuelles.
En France, le CIRAD possède un siège social à Paris et un centre de recherche à Montpellier,
ainsi que quelques stations dans les DOM-TOM.
Les principales activités du CIRAD de Montpellier sont :
- la recherche : en interaction avec les projets du terrain. Le centre de Montpellier fourni
des méthodes scientifiques (analyse des génomes, amélioration des plantes,…),
des techniques (culture in vitro, biologie moléculaire, analyses minérales,…) et
des outils (bases de données, statistiques,…).
- les prestations : mises à la disposition des chercheurs et des agriculteurs des régions chaudes
(identification de maladies tropicales, choix de moyens de lutte, sélection variétale, analyse de bois, aliments,…).
- l’expertise : pour fournir des moyens comme la conception et l’évaluation de projets, ou
encore l’appui aux politiques de recherche agronomique nationales et internationales.
- la formation : le centre accueille plus de 800 chercheurs et techniciens chaque année.
Il est divisé en 7 départements de recherche :
- CIRAD-tera : Territoires, Environnements et Acteurs
- CIRAD-forêt : Forêts
- CIRAD-amis : Amélioration des méthodes pour l’innovation scientifique
- CIRAD-cp : Cultures Pérennes
- CIRAD-ca : Cultures Annuelles
- CIRAD-flhor : Productions Fruitières et Horticoles
- CIRAD-emvt : Élevage et médecine vétérinaire
J’ai été accueilli dans le département Cultures Pérennes du centre de recherche de Montpellier
qui est intégré au parc Agropolis avec 17 autres établissements de recherche et d’enseignement supérieur.
Ce centre regroupe environ 1000 agents, dont prés de 400 chercheurs.
Les filières d’exportation liées aux cultures pérennes sont des éléments moteurs de l’économie de la plupart des pays des zones tropicales humides.
Elles contribuent à intégrer ces pays dans les échanges internationaux.
Elles sont aussi des facteurs de structuration sociale et de développement local.
En alliance avec un large réseau de partenaires, le département des cultures pérennes du Cirad, créé en 1992,
héritier des centres de recherches techniques en zone tropicale humide,
propose des solutions innovantes assurant aux filières concernées un développement durable et respectueux de l’environnement.
Il regroupe 5 programmes de recherche filière :
cacaoyer, caféier, hévéa, palmier à huile, et cocotier qui m’a offert ce stage.
Ils ont pour objectifs :
- l’analyse des mécanismes de fonctionnement des filières et l’aide à la décision de leurs acteurs dans les domaines de la production, de l’organisation de la production et de la transformation primaire des produits bruts;
- la conception de stratégies de production préservant l’environnement : alternatives à la déforestation, cultures associées, protection intégrée des cultures, fumures raisonnées, etc;
- l’analyse des chaînes d’élaboration de la qualité et l’amélioration des opérations de post-récolte, des transformations primaires et des produits finis.

Le cocotier occupe une place importante dans les économies et les systèmes de production de la zone intertropicale,
principalement en Asie, dans le Pacifique et dans les situations littorales et insulaires.
Avant tout villageoise, la culture présente un aspect vivrier avec l’eau, l’amande et la sève,
tout en assurant un revenu régulier aux producteurs au travers de la filière coprah.
Depuis quelques années, le coprah supporte difficilement la concurrence des autres oléagineux tropicaux et même tempérés,
et la filière pourrait disparaître dans les situations les moins compétitives.
Dans ce contexte, le programme Cocotier a choisi d’orienter ses recherches
vers l’amélioration de la productivité de la culture et du revenu du producteur,
la lutte intégrée contre les dépérissements létaux et l’appui à la diversification des usages du cocotier.
Luc Baudouin, membre de ce programme, est mon tuteur au CIRAD.
Il est sélectionneur cocotier. Dans le cadre du réseau Cogent,
il participe à la réalisation d’un kit de marqueurs moléculaires destiné à identifier les cultivars de cocotier.
Ce kit est fondé sur l’utilisation de 14 marqueurs microsatellites avec 600 individus représentant une centaine de cultivars.
- Sources non-Internet :
- Nicolas Marseillac, CIRAD.CP palmier, 2001
L’Institut National de la Recherche Agronomique est un établissement public à caractère
scientifique et technologique placé sous la tutelle du ministère de la Recherche
et du ministère de l’Agriculture et de la Pêche.
Quelques unes de ses missions :
- Mieux nourrir les hommes et de préserver leur santé.
- Gestion de l’espace, des ressources naturelles et de la biodiversité, valorisation des déchets.
- Favoriser l’emploi en identifiant les déterminants de la compétitivité.
- Diversifier les produits et améliorer leur compétitivité et leur qualité.
- Connaître le vivant et développer le génie des procédés : structure et fonction de génomes modèles, génomique et physiologie des organismes, technologies de transformation et de conservation…
Le CBGP, dont le projet a commencé en 1989, a été créé en 1999 par une convention d’Unité Mixte de Recherche (UMR)
cosignée pour 4 ans (1999/2002) par les organismes INRA/IRD/ENSAM/CIRAD.
Le CBGP a pour vocation de comprendre les mécanismes qui régissent les populations d’organismes importants pour l’agriculture,
l’environnement et la santé humaine.
L’objectif finalisé du CBGP est de contribuer à l’amélioration de stratégies de lutte contre les espèces nuisibles
et à l’identification de stratégies de conservation pour des populations naturelles menacées.
Divers types d’organismes constituent le matériel biologique de recherche du CBGP :
des rongeurs, des crapauds, des insectes, des acariens,…
Le CBGP étudie la structuration des populations en fonction des pressions de sélection et des environnements.
L’estimation des flux géniques entre populations est un thème de recherche privilégié,
ne serait-ce que pour prévoir la dissémination de gènes particuliers.
La modélisation est un outil-clé, pour intégrer les différentes échelles d’études
ainsi que pour analyser des scénarios de gestion de manière quantitative.
Un effort particulier est déployé pour intégrer les approches de la génétique et de la dynamique des populations.

Actuellement, le CBGP, dirigé par Yves Gillon,
regroupe 56 agents permanents (32 INRA, 18 IRD, 6 CIRAD) dont 13 HDR (Habilitation à Diriger des Recherches).
En décembre 2000, les 6 équipes du CBGP sont installées sur le Campus international de Baillarguet.
- Biosytématique et écologie (J.Y. Rasplus)
- Génétique des populations en déséquilibre (Jean-Marie Cornuet)
- Biologie et gestion des pullulations (P. Delattre)
- Résistance aux pesticides et gestion des populations (S. Manguin)
- Relations populations-environnement et lutte biologique (Jacques Fargues)
- Modélisation des interactions cultures-ravageurs (S. Savary)
Une UMR fonctionne par mandats de 4 ans, et à la fin de chaque mandat, un bilan global est réalisé.
Si celui-ci n’est pas satisfaisant (problèmes de gestion, organisation, non-respect des impératifs de recherche,…),
la pérennité de l’UMR est remise en cause.
J’ai été accueilli dans l’équipe 2.
Les populations à gérer sont en général celles dont la démographie subit des variations importantes,
introduisant par là-même des déséquilibres au niveau génétique qui rendent inapplicables
beaucoup de méthodes d’analyse en génétique des populations.
 Les efforts de l’équipe 2 se concentrent donc sur le développement de méthodes capables de prendre en compte
les variations démographiques d’une part ou de s’affranchir de l’évolution démographique passée
(génétique des populations en temps réel) d’autre part.
Les efforts de l’équipe 2 se concentrent donc sur le développement de méthodes capables de prendre en compte
les variations démographiques d’une part ou de s’affranchir de l’évolution démographique passée
(génétique des populations en temps réel) d’autre part.
Les recherches incluent des aspects théoriques (développement et tests de méthodes d’analyse)
ainsi que des aspects expérimentaux sur différentes espèces d’auxiliaires (acariens Phytoséides, abeille domestique)
et de bioagresseurs (acariens Tétranyches, crapaud géant, criquet migrateur).
L’équipe 2 est dirigée par Jean-Marie Cornuet qui s’occupe notamment de
l’assignation d’individus à des populations sur la base de leurs génotypes, de la détection génétique des tendances démographiques,
de la génétique évolutive de l’abeille domestique Apis mellifera et de la structuration génétique des populations
de l’acarien Varroa jacobsoni en relation avec la caractérisation de la tolérance des abeilles.
Sylvain Piry, ingénieur d’étude dans cette équipe a été mon tuteur au CBGP.
Il dispose d’une double formation biologie-informatique, ce qui nous a permis de coopérer pour le développement de GeneClass 2.
Il a déjà réalisé les logiciels Bottleneck (1998) et la version précédente de GeneClass (1999).
D’une manière générale, il travaille à l’intégration des aspects informatiques (bases de données, logiciels)
dans les recherches biologiques menées dans les équipes 1 et 2.
- Sources non-Internet :
- Anne Guichard, INRA.CBGP 1, 2001
Action Thématique Programmée
Mon stage s’inscrit dans le cadre d’une Action Thématique Programmée (ATP), proposée par Luc Baudouin (CIRAD.CP) et
Jean-Charles Maillard (CIRAD.EMVT).
Elle s’intitule : Approches biomathématique et biotechnologique pour l’identification génétique
et la gestion adaptée des populations animales et végétales.
Les paragraphes qui suivent sont extraits de la proposition d’ATP par Luc Baudouin le 09/10/2000.
Résumé
Il s’agit de mettre au point un système fiable, économique et discriminant pour identifier
la population d’origine d’un ou plusieurs individus.
Les méthodes actuelles sont insuffisantes lorsqu’on s’intéresse aux animaux ou aux plantes allogames.
Une méthode bayesienne d’indentification sera associée à plusieurs techniques moléculaires dont le multiplexage de microsatellites.
L’efficacité de la démarche sera testée sur 5 modèles animaux et végétaux.
Coopérants, lieux et financement
Les départements CP, EMVT et FORET du CIRAD coopéreront avec l’INRA et le GEVES.
Les lieux de réalisation seront les sites INRA et CIRAD de Montpellier, GEVES à Surgères, l’INRA de Lusignan mais aussi
l’université de Kasetsart à Bangkok (Thaïlande), Port Laguerre à Nouméa (Nouvelle Calédonie) et le CIRAD-FOFIFA de Madagascar.
Le financement sera, sur 3 ans, de 7.6K€ pour l’équipement, 59K€ pour le fonctionnement courant,
25.8K€ pour les frais de missions,
11K€ pour les transports, 21K€ pour les salaires des travailleurs temporaires pour un total de 124K€.
Justification de la demande
L’approche bayésienne a été appliquée à des problèmes d’identification dans des domaines très divers.
Cependant, son application à l’identification des populations est nouvelle (identification d’un échantillon inconnu formé d’un ou plusieurs individus
grâce à une base de données des origines possibles).
L’originalité de la méthode est qu’elle tient compte du nombre limité d’individus observés
et donc de l’incertitude sur les paramètres génétiques.
Ceci est crucial pour l’exploitation de l’information de l’information apportée par les allèles rares.
Il reste à optimiser la méthode et la rendre accessible à travers un logiciel.
Un autre objectif, qui sort du cadre de ce stage, est la mise au point et le multiplexage des microsatellites.
En associant ces méthodes, les participants de l’ATP seront à même de proposer un service attractif
dans le domaine de l’expertise génétique des populations à fécondation croisée.
Ses techniques seront mises en pratique sur le cocotier (pour lequel le CIRAD.CP fournira un kit microsatellites performant),
le cerf rusa (dont l’histoire démographique est très bien connue, ce qui en fait un modèle d’étude unique),
l’Eucalyptus grandis, des lots commerciaux de ray-grass et chez des colonies d’abeilles domestiques.
Cette ATP permet de regrouper des équipes venues d’horizons divers, de l’animal au végétal.
Il permettra une mise en commun des connaissances respectives pour le développement des méthodologies d’identification
et l’interprétation des résultats dans des domaines variés.
L’utilisation, en fin d’ATP, d’un logiciel spécifique assurera le maintien des contacts entre équipes.
Produits attendus, valorisation des résultats
L’ATP débouchera, dans 3 ans, sur plusieurs produits :
- Un logiciel consacré à l’indentification génétique sera développé et couvrira les fonctions suivantes :
- gestion des données.
- identification, conformité et optimisation du choix d’un jeu de marqueurs discriminants.
- Un manuel accompagnera ce logiciel. Une partie sera consacrée à l’utilisation du logiciel,
l’autre à la résolution d’exemples tirés de plantes modèles.
- La constitution de bases de références pour l’identification des populations dans les espèces-modèles et
l’identification des marqueurs les plus discriminants sera un produit scientifique important,
directement valorisable par les participants.
- Des jeux de marqueurs microsatellites multiplexés seront mis au point sur les différents modèles.
- Le rapport final sera conclu par des recommandations sur l’indentification des populations assistées par marqueurs, selon les modèles.
- Enfin, les méthodes développées donneront lieu à des publications scientifiques et feront l’objet d’une présentation sur le Web des savoirs.
Intégration personnelle
Dès le début, il était prévu que je travaille au CBGP. Néanmois, quelques retards ont fait que j’ai passé le premier mois au CIRAD,
où j’ai eu des contacts avec les équipes d’autres stagiaires de l’IUP.GMI. Au cours de ce premier mois, j’ai développé un logiciel
d’assignation de populations n-ploïdes, ce qui m’a permis de me familiariser avec les problèmes d’assignation.
J’ai ensuite été très bien accueilli au CBGP. L’ambiance chaleureuse a largement contribué au bon déroulement de ce stage.
L’équipe, très sympathique, m’a permis de m’intégrer facilement, et par là même de découvrir son fonctionnement, ses objectifs et ses moyens.
J’ai ainsi pu me remémorer mes quelques notions de biologie, apprendre un peu de théorie de la génétique des populations
et comprendre quelques techniques employées en manipulation.
J’ai aussi eu des contacts avec la partie informatique du CBGP que sont Sylvain Piry et l’administrateur réseau Jean-Philippe Melis.
L’intégration au sein de cette équipe c’est très bien passée et a été particulièrement enrichissante,
aussi bien sur le plan humain que professionnel.
Situation de départ
Solutions existantes et besoins
Différents logiciels permettent de faire certains calculs dans le domaine de la
génétique des populations, mais généralement chacun utilise son format de fichier propre.
Leur utilisation n’est pas toujours simple ni agréable, et certaines fonctionnalités manquent.
De plus, ils sont souvent limités à des individus diploïdes (ayant 2 paires de chromosomes).
Il existe aussi plusieurs logiciels faisant des calculs similaires.
Cela est gênant car il y a une perte de temps pour passer d’un logiciel à l’autre,
les algorithmes ne sont pas toujours testés profondément, ou peu explicités,
et les innovations ne sont pas répercutées à toutes les personnes intéressées.
Dans le domaine de l’assignation, il serait donc souhaitable de centraliser
ses calculs au sein d’un même logiciel plus souple et complet.
Cahiers des charges
Ce stage s’inscrit au sein d’un projet sur 3 ans.
Aussi, un cahier des charges a été établit en début de stage indiquant les priorités à atteindre.
Un questionnaire simple et général m’a permis de mieux cerner les demandes.
Cahier des charges initial
Les orientations et priorités m’ont été fournies dans un premier temps par Luc Baudouin (CIRAD.CP cocotier),
sur une vision assez théorique, et avec une grande liberté dans le choix de l’implémentation.
Le début de la liste des priorités était :
- Choix d’une structure des fichiers d’entrées.
- Lecture correcte et sécurisée des données.
- Interface utilisateur avec paramètre d’exécutions, gestion des marqueurs utilisés, gestion des populations.
- Mise en place de l’application 1 : assignation d’échantillons inconnus à une population parmi celles de références.
- Test des individus aberrants
- etc.
De plus, grâce au questionnaire initial, il est apparu qu’un logiciel multi-plateformes (Windows/Linux) et multi-langues serait souhaité.
La priorité devait être la rapidité d’exécution, mais pas au détriment de la facilité d’évolution.
Une compatibilité import/export avec des données de logiciels existants serait très utile.
Le choix du langage de programmation et de la structure de fichiers restaient libres.
Cahier des charges final
L’intégration à l’équipe de l’INRA.CBGP.2 a apporté d’autres impératifs et modifié les priorités :
- Mise en place d’une structure de données efficace et réutilisable.
- Solution permettant de généraliser les algorithmes d’assignation de populations et d’individus.
- Tous les produits doivent fonctionner sous Windows et Linux.
- Lecture et écriture de différents formats de fichiers existants.
- Choix d’une structure des fichiers d’entrées, qui doit être un sur-ensemble des possibilités des autres formats de fichiers supportés,
et accepter les nouvelles fonctionnalités comme la ploïdie variable.
- Evolutivité et compatibilité descendante et ascendante de ce format de fichier.
- Détection et affichage des erreurs de lecture de ces formats.
- Réimplémentation et validation de certains (à terme tous) algorithmes de GeneClass 1 en les généralisant
pour l’assignation de populations et d’individus, et ce quelle que soit la ploïdie. (GeneClass 1 ne traitait que l’assignation d’individus diploïdes).
- Prévoir un système d’installation automatique de GeneClass 2 sur les postes Windows.
- Distributions au moins en français et en anglais.
- Production d’un document d’aide pour développer sur cette structure de donnée.
- Développement d’une version console, avec sortie sur fichier des résultats.
- Orientation vers du calcul distribué pour les calculs de puissances avec la version console de GeneClass 2.
Outils disponibles
Un PC a été mis à ma disposition fin octobre au CBGP. Je l’ai utilisé pour faire les essais sous Linux, mais j’ai surtout travaillé
sur mon PC portable personnel ainsi que sur les machines de Sylvain Piry lorsque nous travaillions ensemble.
Des licences de Delphi 6 et Kylix 2 version professionnelles ont été achetées par le CBGP.
En attendant de les recevoir, nous avons travaillé avec des versions d’évaluation (d’où certaines captures d’écran en anglais dans la suite du rapport).
Techniques utilisées
Borland Delphi 6 est un langage et un environnement de programmation.
Les 10 avantages qui ont influencé ce choix :
- l’efficacité (rapidité) des programmes qu’il compile : c’est un compilateurs des plus efficaces, produisant des exécutables souvent plus performants que les compilateurs C++ (MS Visual C++, gcc, C++ Builder,…).
- la portabilité du code source sous Linux avec Borland Kylix.
- la réutilisabilité aisée du code à travers les composants CLX (fonctionnels sous Delphi 6 pour Windows, Kylix 2 pour Linux, voire C++Builder 5 pour Windows).
- le fait que plusieurs personnes au CBGP connaissent le Pascal et pourront ainsi utiliser les composants CLX.
- la rapidité de développement.
- la clarté du code source (Pascal) pour une maintenance aisée.
- les experts de changements de langue, pour pouvoir distribuer un même programme dans plusieurs langues.
- l’environnement de développement RAD, très agréable et efficace (inspection de code, modèles de code, déboguage,…).
- le même environnement de programmation que Delphi avec Borland Kylix 2 pour Linux, sous des versions “open source” ou “commerciales”.
- le fait que certains programmes aient déjà été faits en Delphi au CBGP.
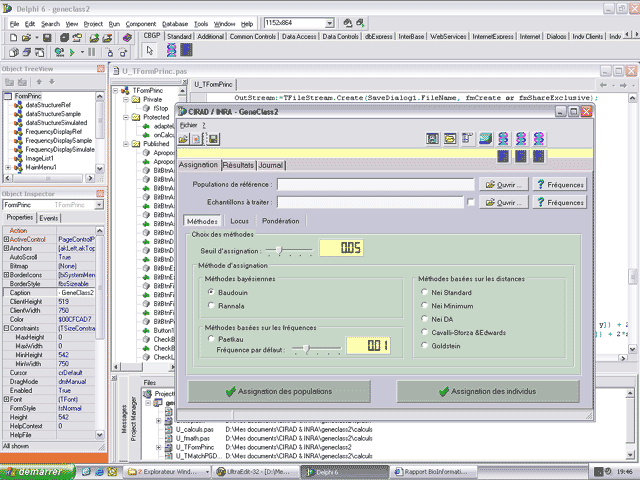
Voici une capture d’écran lors du développement du logiciel GeneClass 2
utilisant les composants CLX qui ont été développés à son intention (onglet CBGP).
Structuration des données fichier avec le XML
Le XML est un format de structuration des données dans un fichier texte.
C’est un standard généraliste qui est en train de s’imposer sur Internet mais aussi pour des données locales et même pour des bases de données moyennes.
Il est directement éditable comme fichier texte, et il existe déjà des outils permettant d’accéder aux données dans la majorité des langages et des plateformes.
Il présente beaucoup d’avantages, entre autres :
- son universalité
- sa flexibilité, en particulier lorsque le format de données doit évoluer
- le contrôle de l’intégrité des données au niveau lexical, grammatical, et éventuellement sémantique.
- l’association qui peut être faite avec des feuilles de présentation, pour afficher les données de diverses manières.
Lecture des autres formats de fichier avec Lex & Yacc
Lex est un générateur lexical, c’est à dire qu’il génère du code permettant de découper du texte en petits morceaux “tokens” selon les règles spécifiées : par exemple un token pourra être un mot ou un nombre.
Yacc est un générateur grammatical, c’est à dire qu’il génère du code permettant d’analyser du texte selon l’ordre des tokens retournés par Lex, et vérifie que les règles grammaticales spécifiées sont respectées.
Lex & Yacc sont donc deux outils, qui, utilisés ensembles, permettent de générer le code source d’un parseur de fichier.
En plus du nouveau format de donnée XML, il est nécessaire de pouvoir lire d’autres formats de fichiers déjà existants.
La lecture d’un fichier dans un format étranger et l’utilisation de ses données (“parsage”) est quelque chose de fastidieux s’il doit être programmé à la main.
La lecture de 7 formats de fichiers nécessaires combinée au fait qu’ils peuvent être au format PC(#13#10), Unix(#10), MAC(#13), PC(#10#13), représentait un gros travail.
Lex & Yacc permettent d’écrire des parseurs rapidement, avec une évolution facile.
De même, il est aisé de changer de langage de programmation.
(Tout cela est néanmoins au détriment de la vitesse de lecture face à un parseur écrit à la main)
C’est pour ces raisons qu’il a été choisi d’utiliser Lex & Yacc.
- Dans un langage propre à Lex, on décrit la lecture lexicale du format de fichier traité, ainsi que les changements d’état (ex: attente de fin de commentaire). Les événements et appels éventuels de fonctions sont des morceaux de code Delphi.
- Dans un autre langage propre à Yacc, on décrit les règles grammaticales. Ce qu’il advient des données lues par Lex est écrit avec des morceaux de code Delphi.
- Lex et Yacc sont alors exécutés, ce qui produit un code source (ici du Delphi) permettant de parser ce format de fichier.
- Le code généré est intégré dans le programme complet.
Quelques modification ont été apportées à dont Lex & Yacc nous avons les sources :
- Lex & Yacc ont été mis à jour pour qu’ils puissent profiter de Kylix et marcher sous Windows ou Linux.
- La librairie de Lex (
lexlib.pas) a été enrichie pour pouvoir lire aussi les formats textes Unix et MAC.
- La librairie de Lex (
lexlib.pas) tire maintenant parti de nouvelles fonctions Delphi 6.
Voir en annexe un exemple de parseur en Lex & Yacc.
NullSoft Install System 1.94 (NSIS) est un outil permettant de déployer des applications pour Windows.
Un script détaillant les fichiers à fournir, les adresses et les menus à créer doit être écrit.
Puis il est compilé avec “makensis” (make NSIS) qui crée un exécutable Windows
avec une interface utilisateur d’installation classique, il y ajoute les fichiers à distribuer compressés.
Les atouts retenus de NSIS sont :
- Sa popularité. C’est un outil assez utilisé, et son interface est connue. (Il a été développé et il est utilisé pour WinAmp.)
- Sa gratuité, même pour des applications commerciales.
- La petite taille des distributions : un seul fichier est généré contenant les fichiers à distribuer compressés en bzip2 et l’interface d’installation qui rajoute environ 36Ko.
- La désinstallation classique, par “ajout/suppression de programmes” sous Windows.
- La simplicité et la puissance de son langage. Presque tout est possible dans le domaine de l’interface d’installation.
Commentaire automatique des sources avec SoftConsult DelphiDoc
SoftConsult DelphiDoc 2.0 est un outil permettant de générer une documentation HTML des sources Delphi avec une navigation hypertexte.
SoftConsult DelphiDoc a été choisi pour :
- Sa gratuité.
- Sa ressemblance avec JavaDoc.
- Ses sorties en HTML.
Models@Home du “Center for Molecular and Biomolecular Informatics of Netherlands” (CMBI)
est un système permettant de distribuer du calcul à plusieurs PCs en réseau (Windows ou Linux).
Pour valider les différents algorithmes d’assignation de GeneClass 2, il est nécessaire de faire des tests de puissance.
C’est à dire qu’il faut simuler un grand nombre de jeux de données différents et comparer pour chacun d’entre eux la qualité d’assignation.
Cela représente une grande quantité de temps de calcul.
Aujourd’hui, les calculateurs sont toujours chers,
alors que la puissance des ordinateurs domestiques s’accroît vite pour un prix en baisse.
Ainsi, pour moins de 1000€, il est possible de monter un PC à prés de 2Ghz avec les périphériques nécessaires
(ex:512 MoDDRam, 60Go disque dur, LAN 1Gbit/s,…).
Le rapport prix/puissance de calcul de ce type de machine est bien plus intéressant que pour des calculateurs spécialisés,
sans parler du coût de fonctionnement.
Aussi, le calcul distribué (clustering) est de plus en plus utilisé : il consiste à partager un gros calcul entre plusieurs PCs.
Models@Home a été choisi pour :
- Sa gratuité.
- Sa facilité de mise en place et d’utilisation.
- Sa facilité d’adaptation aux problèmes à traiter par GeneClass 2.
- Son fonctionnement Windows Linux.
Voir en annexes l’utilisation du clustering.
Rapport de stage en HTML
Le HTML (HyperText Markup Language) est un langage de présentation, permettant, par un jeu de balises comme <BR>
de mettre en forme du texte avec éventuellement des images ou d’autres médias.
Le HTML a été choisi pour écrire et publier ce rapport car :
- c’est un des formats les plus adaptés à la lecture à l’écran.
- il est reconnu sur toutes les plateformes (Windows, Linux,…).
- la diffusion de mon journal d’activité puis de mon rapport en HTML sur Internet a permis à mes tuteurs de suivre l’évolution de mon stage presque quotidiennement.
- les liens hyperTextes enrichissent grandement son contenu, ses citations et facilitent la navigation. Ce rapport en comporte plus de 250 (plus de 800 en comptant DelphiDoc).
- une conversion est possible vers le PDF (Adobe Portable Document Format) pour être imprimée sur papier proprement, sous de nombreuses plateformes.
- l’ATP souhaitant diffuser ses résultats sur Internet, le fait que mon rapport ait été fait en HTML lui donne plus de chances de pouvoir être réutilisé.
- n’importe quel éditeur de texte brut permet de concevoir et de modifier un document HTML
- c’est un des formats les plus légers, à la fois en taille, et en puissance de machine nécessaire pour le visualiser.
- il peut être transformé facilement vers presque n’importe quel autre format (Word, …) alors que le sens inverse ne donne pas de bons résultats.
Ce rapport respecte en général les recommandations du W3C (World Wide Web Consortium),
qui fait référence en la matière, sous la version HTML 4 - (1997, ce qui est assez vieux pour ne plus avoir à se soucier de navigateurs non-compatibles…).
Les feuilles de styles CSS (Cascading Style Sheets) ont été employées pour les détails de présentation comme la couleur, l’alignement,…
(Passage du document en XHTML 1.0 Transitional en juillet 2003, en attente de passage en XHTML 1.0 Strict)
Résultats
Composants CLX, un résultat intermédiaire réutilisable
Les composants sont des objets manipulables graphiquement pendant la conception. Ils permettent une grande réutilisabilité du code.
Le masquage objet prend ici tout son sens, l’utilisateur de ces composants voyant en graphique une bonne partie des méthodes publiques de ces composants.
Pour tous les composants (graphiques et non graphiques), une part du travail de programmation se fait de manière visuelle
dans l’environnement de programmation, notamment la gestion des événements et les liens entre composants.
Ces composants (dont des équivalents existent chez des concurrents de Delphi comme MsVisualC++ ou MsVisualBasic)
sont la méthode actuelle de développement la plus rapide, aisée et fiable.
La CLX (cross-platform component library), qui tend à remplacer la bibliothèque VCL de Delphi, est une bibliothèque de fonctions,
de composants graphiques et non graphiques. Son but est d’être multi-plateforme et efficace.
Elle utilise la bibliothèque QT pour tout ce qui est graphique.
Un code source écrit en utilisant la bibliothèque CLX est portable sans aucune modification de Windows avec Delphi
à Linux avec Kylix et inversement.
Importation dans Delphi ou Kylix des packages de composants CLX
Pour que les composants développés pour GeneClass 2 soient disponibles dans Delphi, Kylix, voire C++Builder, il faut les importer.
Il sont distribués sous la forme de deux packages, contenant chacun un seul composant.
Pour importer d’abord le composant TPGDSStructure qui n’est pas graphique,
il faudra ouvrir (/Fichier/Ouvrir…) au choix :
- pgds_delphi6.dpk : avec Borland Delphi 6 et ultérieur
- pgds_delphi5.dpk : avec Borland Delphi 5
- pgds_delphi4.dpk : avec Borland Delphi 4
- pgds_kylix2.dpk : avec Borland Kylix 2 et ultérieur
- pgds_cbuilder5.bpk : avec Borland C++Builder 5
puis pour le composant TFrequencyDisplay, qui est graphique et utilise directement la CLX,
il faudra ouvrir (/Fichier/Ouvrir…) au choix :
- FrequencyDisplay_delphi6.dpk : avec Borland Delphi 6 et ultérieur
- FrequencyDisplay_kylix2.dpk : avec Kylix 2 et ultérieur
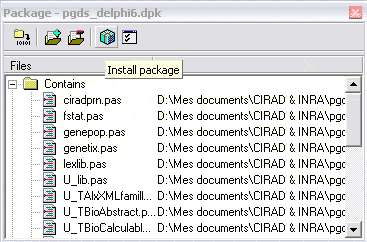
Dans Delphi ou Kylix, cliquez simplement sur installer. Répéter l’opération pour les autres package,
en respectant l’ordre des dépendances. (ex:
TFrequencyDisplay utilise
TPGDSStructure,
donc installer
TPGDSStructure d’abord).
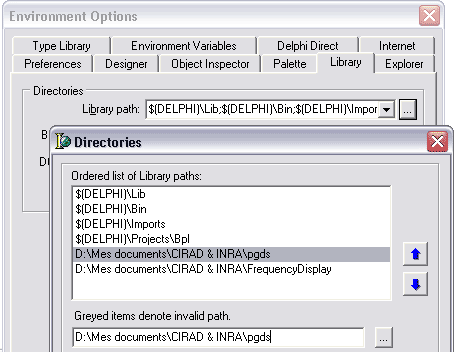
Si vous désirez créer un exécutable autonome, il faudra ajouter aux chemins de recherche de Delphi
(outils/options d’environnement/librairie/chemins des librairies/…) les répertoires contenant les sources
de
TPGDSStructure et de
TFrequencyDisplay.
Si les sources ne vous ont pas été distribuées et que vous avez un package compilé *.bpl cette option n’est pas possible.
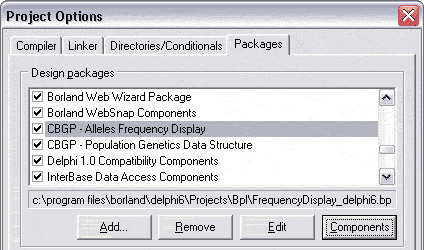
La gestion des packages installés s’effectue dans Projet/Options…/Packages/
Composant TPGDSStructure, structure de données principale
Cette structure objet principale de données, publiée sous la forme d’un composant CLX, est le coeur de mon travail.
C’est elle qui contient les différents parseurs de fichiers. Elle contient après la lecture d’un fichier compatible les différentes
populations avec éventuellement les individus qui les composent. Elle permet aussi de regrouper les populations au sein de groupes.
Toute une gestion des attributs génétiques, tels les allèles pour chaque locus, et des pré-calculs comme les fréquences alléliques,
proportion d’hétérozygotes,… sont fait à son niveau.
Tous les calculs internes ont été masqués et optimisés (ex: recherches dichotomiques, tris QuickSort,…) pour laisser à l’utilisateur de ce composant un outil efficace.
Un gros effort à été fait au niveau de cette structure pour généraliser tout ce qui peut l’être. Les algorithmes d’assignation
qui devaient auparavant être écrits au moins en double pour l’assignation de populations ou d’individus sont maintenant utilisables pour les deux cas.
Ainsi les 2 méthodes d’assignation “Baudouin” et “Rannala” (qui diffèrent peu), et ce pour l’assignation de populations ou d’individus, sont passées de
quatre algorithmes à un seul.
La plupart des algorithmes d’assignation utilisent deux TPGDSStructure pour travailler, l’une servant de référence.
Pour faciliter et optimiser les interactions entre deux TPGDSStructure, un objet TMatchPGDS a été créé.
Pour en connaître plus sur ce composant et voir comment l’utiliser, se conférer aux annexes.
Composant TFrequencyDisplay, informations sur les données d’une TPGDSStructure
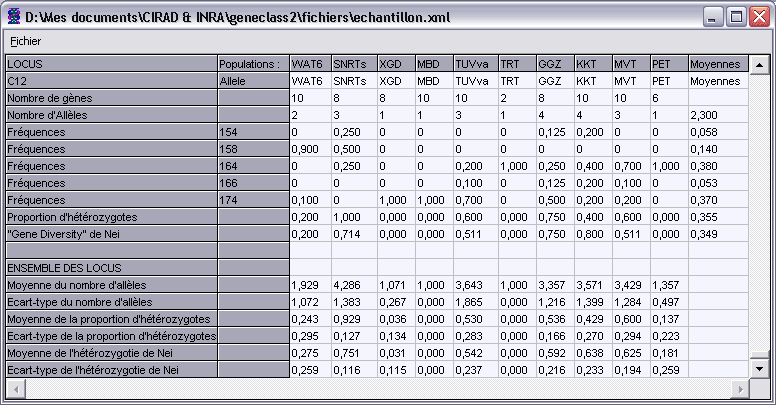
Ce composant graphique est une fenêtre permettant de visualiser une synthèse des données d’une TPGDSStructure.
Pour chaque population et pour chaque locus, il affiche le nombre de gènes relevés, le nombre d’allèles trouvés avec leur fréquence, ainsi que la
proportion d’hétérozygotes et la “Gene diversity” de Nei. Des moyennes sont calculées sur l’ensemble des populations.
Pour l’ensemble des locus sont fournies, pour chaque population, les moyennes et écart-types du nombre d’allèles,
de la proportion d’hétérozygotes, et de l’hétérozygotie de Nei.
Programme minimal utilisant les composants du CBGP
Nous allons voir comment créer rapidement, en utilisant les composants de l’onglet CBGP, un programme très proche du logiciel
Pop100Gene, pour afficher tout ce que sait faire
le composant TFrequencyDisplay, à savoir les fréquences alléliques, …
| Vous allez créer une nouvelle application utilisant la bibliothèque multi-plateformes CLX |
 |
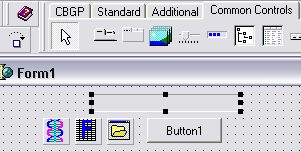
Posez à présent un composant PGDSStructure et FrequencyDisplay de l’onglet CBGP,
un composant OpenDialog de l’onglet Dialogues, une ProgressBar de l’onglet Contrôles Standards.
et un Button de l’onglet Standard |
 |
Puis passez la propriété align de la ProgressBar1 à alTop dans l’inspecteur d’objets. |
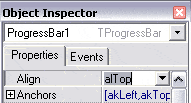 |
Afin que le composant FrequencyDisplay1 sache sur quelle PGDSStructure
il travaille, choisissez PGDSStructure1 comme attribut PGDSStructure pour
FrequencyDisplay1 dans l’inspecteur d’objets. |
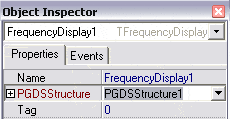 |
Pour insérer le code à exécuter lors d’un clic sur le bouton, double-cliquez sur Button1,
et une procédure vide gérant le clic est crée.
Nous allons maintenant programmer cette méthode, qui permettra, lors d’un clic sur Button1,
d’ouvrir une fenêtre de choix de fichier, de le charger si son format est reconnu dans PGDSStructure1,
et d’afficher les statistiques relatives aux populations et individus qu’il contient avec FrequencyDisplay1. |
procedure TForm1.Button1Click(Sender:TObject);
begin
if OpenDialog1.Execute() then
begin
PGDSStructure1.readFromFile(OpenDialog1.FileName);
FrequencyDisplay1.NewDisplay();
end;
end;
|
Afin d’afficher la barre de progression lors de la lecture du fichier, il faut implémenter l’événement
PGDSStructure1.onDataStructureEvent. Pour cela, double-cliquez sur onDataStructureEvent
dans les événements de l’inspecteur d’objet de PGDSStructure1. |
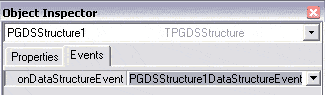 |
| Puis nous allons programmer cet événement, pour lequel une procédure vide vient de se créer automatiquement. |
procedure TForm1.PGDSStructure1DataStructureEvent(
Sender:TObject; const progression:Integer;
out stop:Boolean);
begin
ProgressBar1.Position:=progression;
stop:=False;
end;
|
Un système de compilation conditionnelle a été mis en place pour les composants du CBGP permettant un choix de langue.
Afin que l’instruction de compilation conditionnelle soit répercutée sur le projet en cours ainsi que sur les composants CBGP utilisés,
il faut définir cette variante au niveau du projet. À l’heure de l’écriture de ce rapport, le français (par défaut) et l’anglais sont supportés, mais
il est très aisé d’ajouter d’autres langues (alphabet latin).
Il faut penser à reconstruire (build) complètement le projet après cette modification.
|
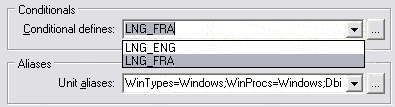 |
C’est fini ! Vous n’avez plus qu’à sauver le projet puis l’exécuter.
Cette opération est identique sous Kylix 2 et donne le même programme sous Linux.
|
Programme minimal (ZIP 1.47Ko)
|
Utilisations avec GeneClass 2

Le logiciel GeneClass 2 est basé sur ce même programme minimal qui a été enrichi entre autres de nombreux algorithmes d’assignation,
ainsi que d’une interface graphique développée permettant de bénéficier pleinement des fonctionnalités de la structure de données TPGDSStructure.
L’ajout d’algorithmes peut être fait au programme minimal précédent pour tester de nouvelles idées, ou encore des variantes des
algorithmes déjà implémentés dans GeneClass 2.
Voir pour cela “Programmation sur la structure de données” en annexes.
GeneClass 2, programme d’assignation
GeneClass 2 est donc un programme utilisant plusieurs composants TPGDSStructure et
permet de faire des assignations d’individus ou de populations à des populations de références, et ce quel que soit la ploïdie
des individus.
Cette nouvelle version de GeneClass est attendue car elle permettra de travailler sur des individus n-ploïdes,
d’assigner des populations complètes, et de travailler sur des plus gros effectifs. Suite à la demande d’un utilisateur qui travaille
avec une base de données de 50000 poissons, GeneClass 2 a été testé avec succès sur des calculs impliquant un
demi-million d’individus.
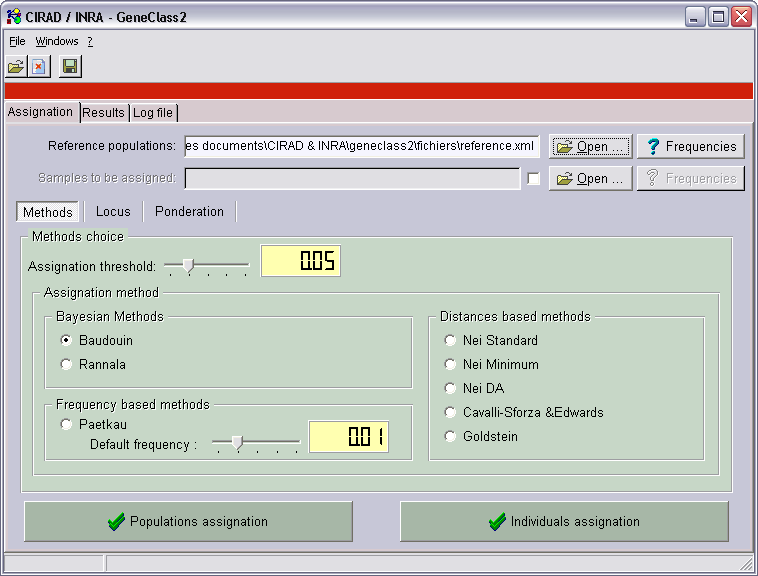
Utilisation
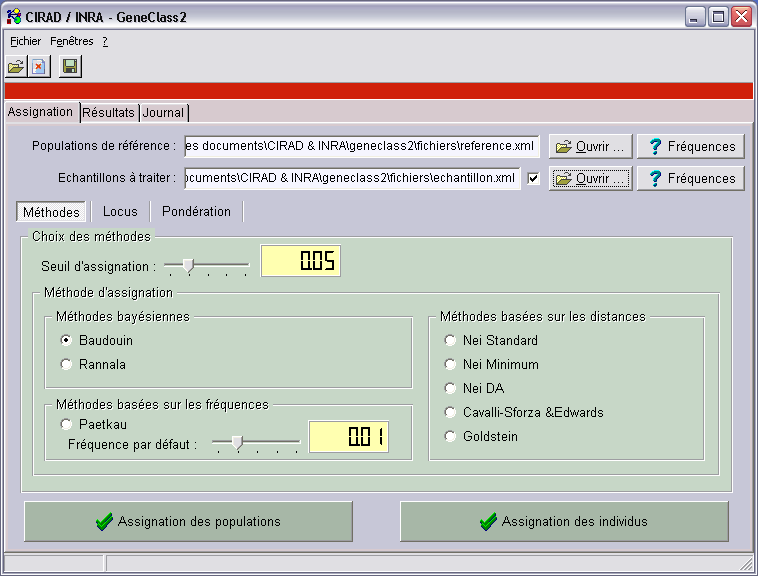
Après l’ouverture de GeneClass 2, il faut ouvrir les fichiers de données. Pour ça, cliquer sur un des boutons “ouvrir”, pour charger
soit un fichier de référence, soit un fichier des échantillons à assigner.
Les boutons “fréquences” affiche une fenêtre TFrequencyDisplay.
Le seuil d’assignation correspond à la valeur minimale en dessous de laquelle les assignations n’apparaissent pas dans les scores.
Il reste alors à choisir une des méthodes d’assignation.
Puis on clique sur “assignation des populations” pour assigner toutes les populations du fichier des échantillons,
ou sur “assignation des individus” pour assigner tous les individus de toutes les populations du fichier des échantillons.
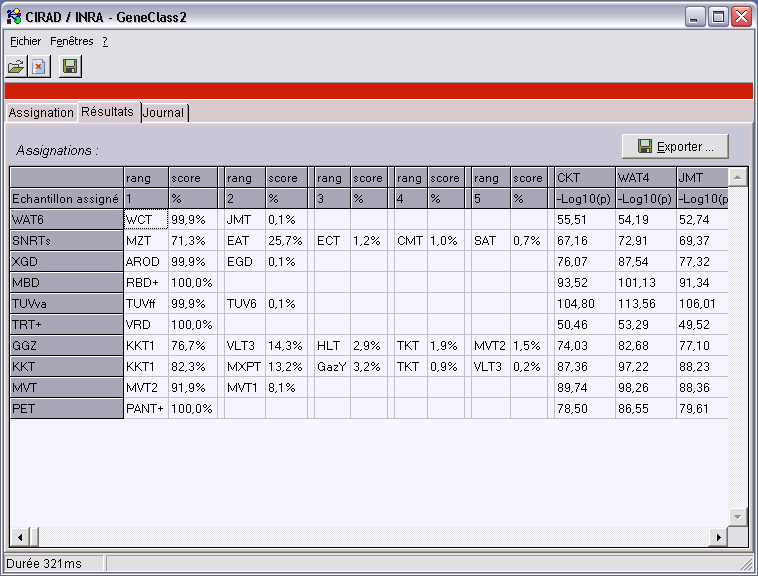
Le changement d’onglet est automatique et il s’affiche une grille des résultats de l’assignation.
- Dans la colonne de gauche se trouvent les noms des populations ou des individus assignés.
- Pour chaque échantillon, on a d’abord une première partie contenant au maximum les 5 meilleures assignations (rang),
avec une valeur corrigée en % (score). La somme des scores fait 100% pour les méthodes bayésiennes et de fréquences.
- Puis on a les valeurs exactes (ou -Log10 de la valeur) obtenues pour chacune des populations de référence du fichier de référence.
- Le bouton “Exporter…” permet de sauver les résultats au format .csv qui peut être ouvert dans un tableur comme Excel,
ou dans une base de données comme Access s’il y a trop de populations.
- Dans la barre d’état s’affiche le temps de calcul. Ici 321ms pour assigner 10 populations (46 individus de 14 locus) à 109 populations de référence (546 individus de 14 locus).
Il est possible aussi de faire de l’auto-assignation. Pour cela, il faut charger uniquement le fichier de référence, ou décocher la case des échantillons.
Dans ce cas, lors de l’assignation d’un individu, cet individu est retiré de sa population qui est recalculée (fréquences alléliques,…).
On parle de “leave-one-out”. On fait cela pour que l’individu n’influence pas l’assignation. En effet, l’individu aurait plus de chances d’être
assigné à la population à laquelle il appartient car il modifie selon son modèle les fréquences allèliques, hétérozygotie, etc. de sa population.
N.B : pour faire de l’auto-assignation sans “leave-one-out”, charger deux fois le même fichier dans référence et échantillons.
Voici un exemple d’auto-assignation “leave-one-out” dans GeneClass 2 version anglaise :
GeneClass 2 version console
Une version console (Windows et Linux) de GeneClass 2 a été développée,
en particulier pour faire des tests de puissances des algorithmes d’assignation.
Usage:
geneclass2cl [--logFile <logFile>] --refFile <referenceFile>
[--sampFile <samplesFile>] [--output <outputFile>]
[--threshold <threshold>] [--method <methodName>]
[--paetkauFreqDef <freq>] [--assignIndividuals]
geneclass2cl [-l <logFile>] -r <referenceFile> [-s <samplesFile>]
[-o <outputFile>] [-t <threshold>] [-m <methodName>]
[-f <freq>] [-i]
<threshold>=0.5|0.05|0.005|0.0005|0.00005|0.000005
<freq>=0.1|0.01|0.001|0.0001|0.00001
<methodName>=baudouin|rannala|paetkau|neistandard|neiminimum|
neida|cavallisforza|goldstein
Algorithmes d’assignation
GeneClass 2 utilise différents algorithmes d’assignation. Leurs résultats ne sont pas forcément identiques mais les tendances
générales doivent concorder. Pour déterminer quels sont les meilleurs algorithmes dans différentes situations, il faut attendre
les résultats des tests de puissances.
Des comparaisons ont déjà été faites par Cornuet et al. dans “New methods employing multilocus genotypes to select or exclude populations as origins of individuals”, Genetics vol.153:1989-2000.
Méthodes bayésiennes Baudouin et Rannala
Ces deux formules bayésiennes ont été généralisées avec un même algorithme, dont seul un paramètre diffère.
Cette formule a été aussi généralisée pour l’assignation d’individus ou de populations, quelle que soit la ploïdie.
Il semble, avant vérification, que la variante de Luc Baudouin soit moins sensible aux erreurs apportées par l’incertitude liée
aux populations de peu d’individus (<5). En d’autres termes, la variante Rannala semble plus optimiste quand à la précision
et l’exactitude des fréquences allèliques des populations.
LnGamma : logarithme de la fonction Gamma, avec Gamma(x)=(x-1)!
M : echantillon testé
N : échantillon de référence
alpha : paramètre de la loi de Dirichelet
alpha = 1 selon Rannala
alpha = k selon Baudouin
k : nombre d’allèles
h : allèle courant
mh : nombre de gènes de l’allèle h dans l’échantillon testé
nh : nombre de gènes de l’allèle h dans la référence
Ln(Pr(M/N)) = LnGamma(m+1)
+ LnGamma(n+alpha)
+ Somme(h=1 à k)[LnGamma(mh+nh+alpha/k)]
- Somme(h=1 à k)[LnGamma(mh+1)]
- Somme(h=1 à k)[LnGamma(nh+alpha/k)]
- LnGamma(m+n+alpha)
on a alors la probabilité d’assignation Pr(M/N) = Exp(Ln(Pr(M/N))).
- Sources :
- Rannala, B. and J. L. Mountain, 1997 Proc. Natl. Acad. Sci. USA 94: 9197-9221
Méthode Paetkau basée sur les fréquences allèliques
La formule de David Paetkau a été généralisée pour l’assignation d’individus et de populations, quelle que soit la ploïdie par Jean-Marie Cornuet.
La fréquence par défaut, visible sur GeneClass 2 correspond à la fréquence que l’on donne à un allèle lorsque celle-ci n’est pas calculable.
Il semble que la méthode Paetkau donne toujours des résultats un peu moins bons que ceux des méthodes bayésiennes.
D’un autre côté, son calcul est toujours plus rapide.
LnGamma : logarithme de la fonction Gamma, avec Gamma(x)=(x-1)!
M : echantillon testé
k : nombre d’allèles
h : allèle courant
mh : nombre de gènes de l’allèle h dans l’échantillon testé
ph : fréquence de l’allèle dans la pop de référence
TIRAGE MULTINOMIAL :
Ln(Pr(M/N)) = LnGamma(m+1)
- Somme(h=1 à k) [LnGamma(mh + 1)]
+ Somme(h=1 à k) [mh * Ln(ph)]
on a alors la probabilité d’assignation Pr(M/N) = Exp(Ln(Pr(M/N)))
- Sources :
- D.Paetkau, W.Calvert, I.Stirling, C.Strobeck: Mol.Ecol. 4:347-354
Méthode basées sur les distances
Je n’ai pas beaucoup participé à l’implémentation des méthodes basées sur les distances.
De plus, les résultats semblent, avant les tests de puissances qui s’imposent,
moins intéressants que ceux fournis par les méthodes précédentes.
La méthode de Goldstein sort particulièrement du lot. C’est la seule qui utilise les écarts de longueur des allèles,
mais les résultats ne sont pas très convainquants.
- Sources :
- N.Takezaki, M.Nei: 1996 Genetics 144: 389-399
Conclusion
Ce stage a représenté une grosse masse de travail.
Le projet GeneClass 2 aura nécessité plus de 14000 lignes de code Delphi, sans compter les commentaires, les programmes de distribution,
les variantes de langues, de plateforme, de version (pour les packages sous Delphi 4,5,6 Kylix 2 et C++Builder 5)
pour un total d’environ 350000 caractères.
Les différents outils choisis se sont révélés satisfaisants.
Les besoins et contraintes apportées par les différentes personnes impliquées dans ce projet ont été dans l’ensemble satisfaits.
L’emploi du temps de l’ATP est, pour cette partie au moins, respecté.
GeneClass 2 est en cours de validation, avec des tests de puissance faits en clustering, notamment en ce qui concerne la correction des données manquantes.
La méthode d’assignation imaginée Luc Baudouin va faire l’objet d’une
publication dans une revue scientifique.
Cette publication permettra la diffusion de GeneClass 2 dans la communauté scientifique concernée.
Des variantes et ajouts seront faits, en particulier sur l’exclusion d’individus (tests de hors-type, populations simulées).
Des outils permettant cette évolution ont déjà été prévus au niveau de la structure de données.
- Mise à jour de novembre 2003 :
- Geneclass 2 est disponible sur la page des logiciels du CGBP.
- Mise à jour de octobre 2004 :
- Publication de GENECLASS2: Un logiciel pour l’assignation génétique et la détection de migrants de première génération par l’équipe du CBGP.
Voir aussi les annexes.
Alexandre Alapetite